
Randomized controlled trials (RCTs), particularly in phase 3, are the preferred sources of evidence for drug approval and reimbursement decisions. Moreover, phase 3 trials are at the top of the evidencebased medicine pyramid, thus representing the gold-standard for judgements about the efficacy and safety of new treatments and their worth in clinical practice. Nevertheless, regulatory and clinical decisions sometimes need to be made on evidence of lower level, such as that provided by phase 2 trials, particularly in oncology. As a matter of fact, nearly one-third of anticancer drug approvals by the US Food and Drug Administration (FDA) between 1992 and 2017 were accelerated approvals, one of the
four types of expedited programs offered by that Agency.1,2
In addition to their potential role as sources of evidence for regulatory and clinical decisions, phase 2 trials play a key role in go/no-go decisions within biotech and pharmaceutical companies. Yet, the transition between phases 2 and 3 is the one associated with the lowest success rate in drug development, and oncology seems to do even worse than other specialties in this respect (32% vs 41% for non-oncology indications).3 This high rate of failure implies that very promising results seen in phase 2 are often not replicated in phase 3; this is a facet of the “replication crisis” that has become widely advertised in the medical literature.4 Why do so many phase 3 trials fail?
Table 1 provides some of the many reasons why positive results from a clinical trial are not replicated. When discussing phase 2 trials, here we refer to their randomized variety; single-arm trials, event though representing a large fraction of accelerated approvals,1 are not as reliable as a source of evidence and cannot properly be qualified as “positive” or “negative”, given the lack of a formal control group and the potential for selection bias, whereby nominally good results may well be due to enrolment of a more favorable population.
Table 1. Some reasons for the “replication crisis” in clinical research.
| Category | Reasons |
| Biological/clinical | Mechanism of action not the one initially expected |
| Differences between assessed populations | |
| Different meaning and implications of different endpoints | |
| Pharmacological | Inadequate dosing or understanding of dose-activity relationship |
| Unfavorable therapeutic window between toxicity and activity | |
| Operational | Data integrity issues |
| Noncompliance with the protocol | |
| Statistical | Play of chance (may overlap with other reasons) |
| Insufficient power (small sample size) | |
| Missing data | |
| Incorrect methodology and “P-hacking” (emphasizing P-values < 0.05) | |
| Regression to the mean | |
| Cultural/social | Pressure to publish |
| Publication bias |
Although any or many of the reasons shown in Table 1 might be present in any given case, one specific reason alone is sufficient to explain, if not “why so many phase 3 trials fail”, at least “why we should not expect phase 3 trials to replicate positive results from phase 2 trials”, at least on average. This reason, characterized in Table 1 as “regression to the mean”, has also been called “the winner’s curse” in a different context, namely that of auctioned items, where the winning bid tends to exceed the intrinsic value of the item (see Figure 1).
Figure 1. Graphical representation of the “winner’s curse”.
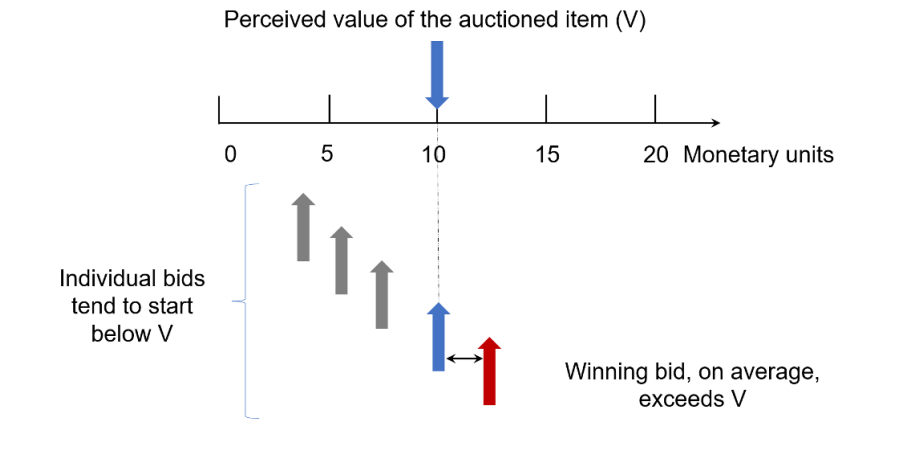
Regression to the mean was first described by Francis Galton almost 140 years ago, based on his empirical finding that male adults were closer to average height than their fathers. In other words, tall people tend to have children who are also tall, but less so—in comparison with population averages— than themselves, and vice-versa for short people. The same mechanism often underlies the frequent finding in clinical medicine that a mildly abnormal laboratory test, if repeated, yields a normal result.
Regression to the mean can be easily understood if one considers that any observation subject to variability (such as the result of a laboratory test or a phase 2 trial) is in fact an individual representative of a population of similar results; such a population has a distribution that is often bell-shaped, with extreme results on either side of the average being relative rare.
In the case of phase 2 trials, the extreme results would be a trial significantly favoring control or one favoring the experimental agent; the former would not lead to a follow-up phase 3 study, but often to discontinuation of the development program. But a phase 2 trial significantly favoring the experimental agent—the one that will often lead to a phase 3 trial—is likely to be followed by an RCT with results that are closer to the null hypothesis of no treatment effect. This is simply due to regression to the mean.
Given the central role of RCTs for regulatory and clinical decisions, the fact that a “positive” trial may be followed by a negative one, even if both have very similar designs, is reason for concern, and one that suggests that accelerated approvals are at times “false positives”. Although this does not appear to be too frequent, of 56 accelerated approvals that have fulfilled their post-marketing requirement, 51 had their benefit confirmed, but five had the product withdrawn from the market, in three cases due to lack of confirmation.1
From the preceding discussion, it is fair to conclude that some of the observed results from early positive trials are exaggerated in comparison with “true results”, the latter being an ideal notion representing the true effect of a treatment. The ratio between the observed effect and the true effect has been called “the exaggeration ratio”.6
Although the true effect cannot be known with certainty, it can be replaced by the treatment effect contained in a large database of clinical trials from the literature, and a website has been built that allows estimation of the exaggeration ratio likely to be present in a phase 2 trial.7
In the discussion about positive and negative RCTs, one must also consider the meaning of “statisticalsignificance”. This is particularly relevant when drug development is done under a frequentist statisticalframework. Historically, this has been the most common framework adopted in clinical research, allowing tests of the null hypothesis of no difference between two treatments. The P-value obtained from such a test indicates the probability of observing a treatment effect at least as extreme as that observed if there were no true treatment effect. By historical convention, a P-value that is less than 0.05 is considered as “statistically significant”, but this is no guarantee that a study is truly positive, nor is it a
direct measure of the treatment effect.
In order to circumvent some of the limitations of the frequentist framework with regard to go/no-go decisions at phase 2, one may use a Bayesian framework, which can help to correct the exaggeration ratio and yields probabilities related to the plausibility of the treatment effect.6,8 This framework can use the results from an early trial in a manner that allows moving from the probability indicated by the Pvalue—namely, the probability of the observed data, given the null hypothesis—to a more relevant one in this setting, i.e., the probability of the null hypothesis, given the observed data.
By looking at the probability of the null hypothesis, or, conversely, that of any potential treatment effect, developers will be able to match their optimism to more realistic probabilities contained in the data from phase 2 trials, thus being best equipped to decide whether or not to proceed to phase 3.

References:
1. Beaver JA, et al. A 25-Year Experience of US Food and Drug Administration Accelerated Approval of Malignant
Hematology and Oncology Drugs and Biologics: A Review. JAMA Oncol 2018;4:849-856.
2. U.S. Department of Health and Human Services. Food and Drug Administration. Guidance for Industry. Expedited Programs for Serious Conditions – Drugs and Biologics, May 2014.
3. Grignolo, et al. Phase III Trial Failures: Costly, But Preventable. Applied Clinical Trials 2016; 25:36-42.
4. Ioannidis JP. Why most published research findings are false. PLoS Med 2005;2(8):e124.
5. Capen, et al. Competitive Bidding in High-Risk Situations. J Petrol Tech 1971;23:641-653.
6. Van Zwet E, et al. A Proposal for Informative Default Priors Scaled by the Standard Error of Estimates. The American Statistician 2022;76:1-9.
7. https://vanzwet.shinyapps.io/shrinkrct/.
8. Benjamin DJ, et al. Three Recommendations for Improving the Use of p-Values. The American Statistician 2019;73:sup1:186-191.
